

Proteins underpin every biological process in an organism. Each protein has its own unique 3D shape, which defines how it works and what it does. To date, scientists have discovered more than 200 million different types of protein, with this number ever-increasing. However, before this year we had no effective way of predicting the 3D shape of most of these proteins given its 1D amino acid sequence.
Google-funded British artificial intelligence company DeepMind made headlines this year after the company’s researchers created a model which could accurately predict the shape of various proteins after they had folded [1]. This has been a Grand Challenge for over 50 years, and DeepMind’s algorithm ‘AlphaFold 2’ is seen by many as one of the biggest advancements in structural biology and artificial intelligence in the past two decades:
“This computational work represents a stunning advance on the protein-folding problem, a 50-year-old grand challenge in biology. It has occurred decades before many people in the field would have predicted. It will be exciting to see the many ways in which it will fundamentally change biological research.” - Professor Venki Ramakrishnan, Nobel Laureate and President of the Royal Society.
A bit about protein folding
Proteins are made up to 20 different types of amino acids, where interactions between these acids lead to the protein folding and thus creating a 3D shape. As you can imagine, with hundreds of millions of proteins, this yields almost an infinite number of possibilities – 10300 in fact (Levinthal, 1969).
The misfolding of proteins has been said to be the underlying cause of many diseases, including Alzheimer’s disease, Parkinson’s disease and cystic fibrosis (Chaudhuri, Paul, 2006). Consequently, scientists have spent years trying to figure out a way to compute a protein’s shape just by using the chain of amino acids, however, previous methods have proven to be very expensive and time-consuming. The most popular and accurate method used before the advent of AI-based solutions was to use X-ray crystallography, which costs around $120,000 per protein and takes 1 year to determine the structure, according to an estimate from the University of Toronto.
CASP
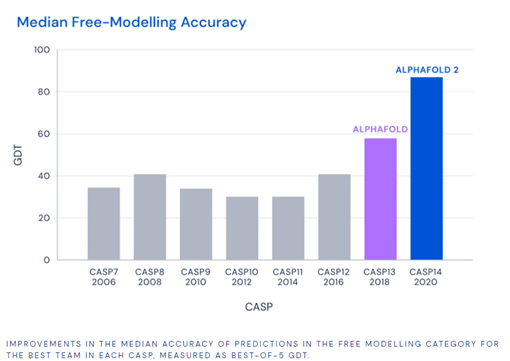
In 1994, Biological Researcher John Moult launched the biennial competition called CASP (Critical Assessment of Structure Prediction), where entrants receive amino acid sequences for 100 proteins with unknown structures. Scores above 90 (scale is 0 to 100) are seen as a viable solution to the problem. Results of the competition are measured by the Global Distance Test, which essentially measures the percentage of predicted amino acid residues within a certain threshold distance from the correct position.
AlphaFold 2
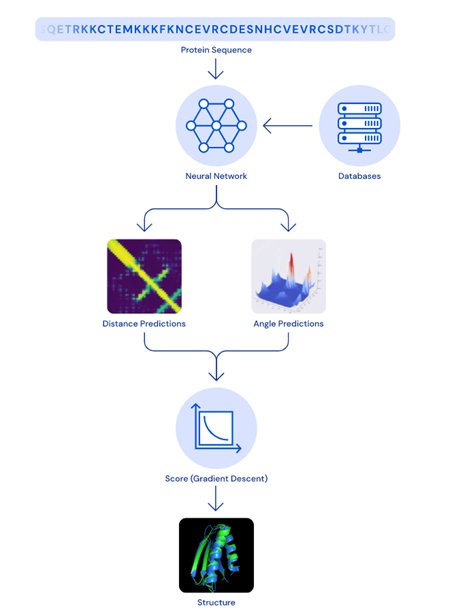
The model that won this year’s CASP competition, AlphaFold 2, uses deep learning to predict the 3D shape of proteins. While a peer-reviewed paper has not yet been released, it is an extension of the previous model, AlphaFold 1. DeepMind maintains that a folded protein can be thought of as a ‘spatial graph’. The residues of the amino acids are the nodes of the graph, and the edges connect the residues in proximity. The latest version of AlphaFold comprises of an attention-based neural network system that interprets the structure of said graph, while reasoning over the implicit graph that it is building.
This process then uses multiple iterations to determine the structure of a protein through its predictions, operating over amino acid residue pairs and evolutionary related protein sequences. Scores are then optimised by using gradient descent, an efficient way to find the most accurate prediction. A key advantage of this model when compared to traditional methods is that the structure may be determined within a matter of days. Additionally, AlphaFold can predict which parts of each predicted protein structure are reliable using an internal confidence measure.
Alphafold 2 was able to achieve an accuracy of 92.4, measured by the median GDT score across all target proteins, outshining the competition and DeepMind’s own previous models. The root mean squared deviations of errors was 1.6 Angstroms, similar to the width of an atom (DeepMind, 2020).
Real-world impact
AlphaFold 2 has brought society one step closer to further understanding biological structures. This is a prime example of artificial intelligence at the centre of solving a Grand Challenge. AI can accelerate the drug discovery process, open up opportunities to create enzymes to break down plastic in the ocean, find quicker and better cures for diseases or even create a structure to capture carbon in the atmosphere.
However, there is still substantially more work to be done. Further to understanding the way a protein folds, scientists may also benefit from understanding how these proteins react with DNA and other small molecules and how we can determine the location of all amino acid side chains. AlphaFold demonstrates an excellent foundation for further innovation via the means of AI in the field of biological research.
References
[1] Sample, Ian., 2020. DeepMind AI cracks 50-year-old problem of biology research. The Guardian, available at: https://www.theguardian.com/technology/2020/nov/30/deepmind-ai-cracks-50-year-old-problem-of-biology-research
[2] Jumper, J. et.al. High Accuracy Protein Structure Prediction Using Deep Learning. In Fourteenth Critical Assessment of Techniques for Protein Structure Prediction (Abstract Book), pp. 22-24, 30 November - 4 December 2020. Available at: https://predictioncenter.org/casp14/doc/CASP14_Abstracts.pdf
[3] Levinthal, C., 1969. How to Fold Graciously. Mossbauer Spectroscopy in Biological Systems: Proceedings of a meeting held at Allerton House, Monticello, Illinois. Available at: https://web.archive.org/web/20110523080407/http://www-miller.ch.cam.ac.uk/levinthal/levinthal.html
[4] Chaudhuri TK, Paul S. Protein-misfolding diseases and chaperone-based therapeutic approaches. FEBS J. 2006 Apr;273(7):1331-49. doi: 10.1111/j.1742-4658.2006.05181.x. PMID: 16689923.
[5] DeepMind, 2020. A schematic of the architecture of the AlphaFold system predicting structure from protein sequence. Available at: https://deepmind.com/blog/article/AlphaFold-Using-AI-for-scientific-discovery
[6] DeepMind, 2020. AlphaFold: a solution to a 50-year-old grand challenge in biology. Available at: https://deepmind.com/blog/article/AlphaFold-Using-AI-for-scientific-discovery